RNA-seq workflow¶
This workflow is used for RNA-seq and RNA-seq-like analysis (like euRNA-seq, RIP-seq or small RNA-seq).
This workflow can use references created by the references workflow with no need to run the references workflow separately. This workflow performs the following tasks:
Builds a HISAT2 index
Builds a salmon transcriptome index
Downloads a GTF annotation
Converts the GTF to refflat format
Trims reads with cutadapt
Aligns with HISAT2
Runs FastQC on raw, trimmed, and aligned reads
Aligns reads to rRNA using bowtie2 to evaluate rRNA contamination
Counts reads in genes with featureCounts
Runs dupRadar and preseq to assess library complexity
Checks for evidence of cross-contamination using fastq_screen on multiple configured genomes
Assesses transcript coverage with Picard CollectRnaSeqMetrics
Builds bigWigs (optionally strand-specific) created from BAM files
Optionally merges bigWigs as defined by config
Aggregates QC results using MultiQC. Includes custom tables for library sizes and rRNA contamination
Runs comprehensive downstream analysis including QC and differential expression in R. See section below for more details.
Constructs and uploads a track hub of scaled coverage bigWigs for each sample that can be viewed in UCSC Genome Browser
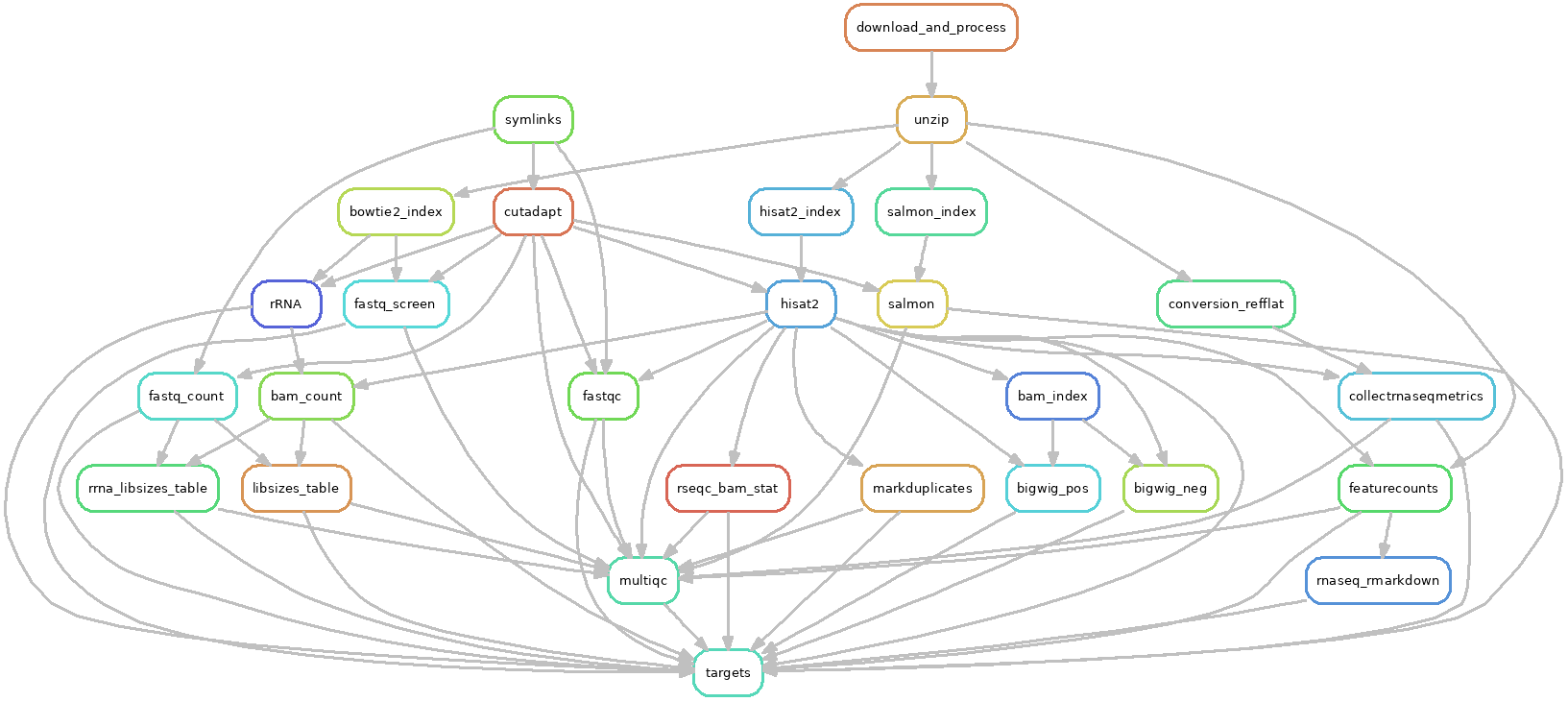
The DAG of jobs looks like this:
Downstream analysis¶
This is performed in an RMarkdown file (rnaseq.Rmd) that uses DESeq2
for differential expression analysis, along with diagnostic plots,
exported tables of differentially expressed genes for each comparison of
interest, gene patterns analysis for finding coexpressed genes and downstream
functional enrichment analysis using clusterProfiler. This file is run and
rendered into an output HTML file. See RNA-Seq downstream analysis for more details.