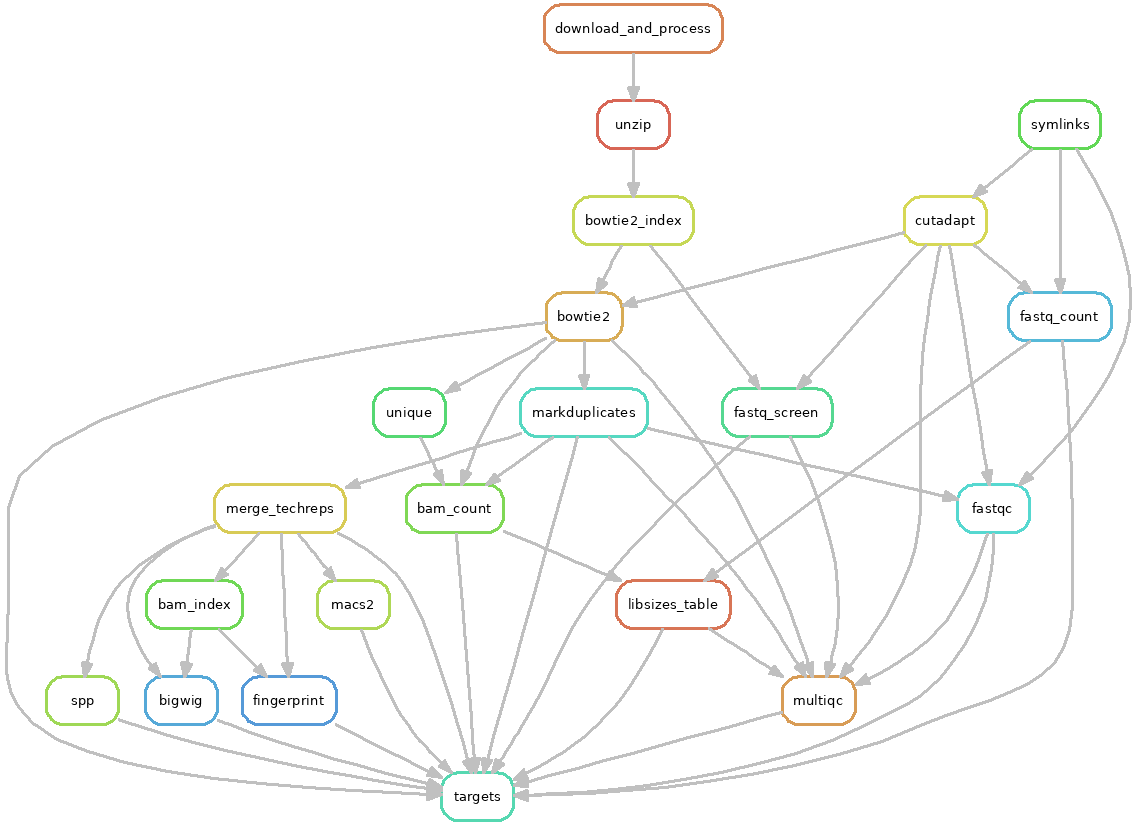
ChIP-seq workflow¶
The ChIP-seq workflow starts with raw FASTQ files and performs various QC steps. It aligns and prepares BAM and bigWig files, performs peak-calling, and combines everything together into a track hub for visualization.
Specifically, the workflow does the following:
trims reads with cutadapt
maps reads with Bowtie2
runs FastQC on raw, trimmed, and aligned reads
Removes multimappers (samtools) and duplicates (Picard MarkDuplicates)
performs fastq_screen on multiple configured genomes to look for evidence of cross-contamination
QC aggregation using MultiQC, along with a custom table for library sizes
merges technical replicates and then re-deduplicates them
creates bigWigs from unique, no-dups BAM files
optionally merges bigWigs to create one signal track for all replicates
runs deepTools plotFingerprint on grouped IP and input for QC and evaluation of enrichment
calls peaks using macs2, spp, and/or sicer, with support for multiple peak-calling runs using different parameters to assist with assessing performance and to help make decisions for downstream analysis
optionally runs a template diffBind RMarkdown file used for differential binding analysis
converts BED files into bigBed (or bigNarrowPeak where possible)
builds and optionally uploads a track hub of bigWigs and bigBeds to visualize peak-calling in UCSC Genome Browser
To configure a ChIP-seq experiment, see Config YAML.